论文名:Normalization and de-noising of scHi-C data with BandNorm and scVI-3D
introduction
主要使用了两种方法,BandNorm和scVI-3D。分别解决了scHi-C数据的normalization和de-noising问题。我们关心的主要的还是scVI-3D模型。
- 数据
Hi-C的数据天生的具有一些bias和noise,比如systematic genomic distance bias,这些噪声由测序的实验引起,很难改进,给下游的分析任务带来很大的困扰。而single cell数据相对于集群细胞的数据而言,又天生的具有稀疏性和噪声。相对于其他的single cell数据,scHi-C数据由于使用了基因组的绝对坐标作为基本的研究单元,所以更加的稀疏。
- 方法
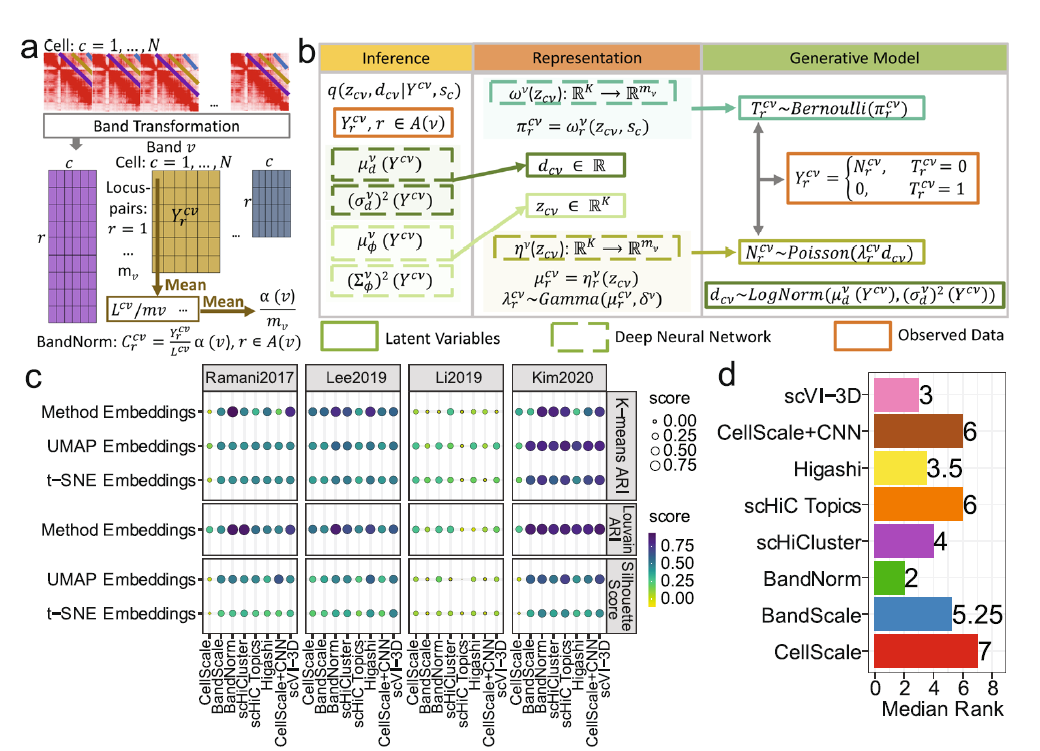
fig-1a介绍了bandnorm方法。在进行bandnorm之前,首先将接触矩阵拆分成band-specific cell 和 基因座矩阵(感觉意思就是把数据拆成每个细胞)。然后再每个细胞的每条染色体上分别进行bandnorm,将原始的接触系数$Y_{r}^{cv}$(r:基因座;v:Band,感觉意思就是bin后的接触矩阵的坐标;c:cell)。bandnorm的公式是:
fig-1b介绍了scVI-3D模型。将$Y{r}^{cv}$投射为隐层变量$z{cv}$…说是让看后边的详细介绍,最关心的就是这一部分,但是这部分最语焉不详。
fig-1c和fig-1d介绍了几种方法的对比,CellScale, BandScale, BandNorm, scHiCluster, scHiC Topics, Higashi, CellScale+CNN, scVI-3D。分别在Ramani2017, Lee2019, Li2019, Kim2020数据集上进行了测试。不是我们关心的部分。
接下来直接跳到论文的第23页看他的scVI-3D模型。
scVI-3D模型
scVI-3D模型将基因座之间的交互频率建模为从零膨胀负二项分布(zero-inflated negative binomial distribution)中得到的采样,同时考虑library size和batch effect。
整体上跟用于scRNA-seq的scVI差距不大,模型的本质是一个VAE模型。原始的VAE模型中认为变量服从高斯分布,使用神经网络学习$\mu和\sigma$。这里的scVI认为输入的数据服从的是零膨胀负二项分布,使用神经网络学习$\theta, r和p$。
零膨胀负二项分布
在介绍零膨胀负二项分布之前,首先介绍一下为什么要使用这样一个分布对交互频率进行建模。之前介绍过,single cell数据天然的具有很高的稀疏性,而且,由于测序数据事实上就是基因座之间的交互频率,这些数据的取值必然是非负整数。很自然的,我们就可以想到使用Poisson分布对scHiC数据进行建模。
- 泊松分布
泊松分布的概率密度函数如下所示:
使用泊松分布描述scHiC数据的问题在于,对于一个泊松分布来说,他的期望和方差是相等的,即$E(X) = Var(X) = \lambda$。但是对于实际的单细胞数据而言,随着基因的表达水平的越高,其样本方差和样本均值的差就越大。即所谓的过度分散性。所以使用泊松分布是不合时宜的。参考scRNA-seq的一些相关解释。引入Gamma分布。
- Gamma分布
Gamma分布的概率密度函数如下所示:
Poisson分布的$\lambda$是不变的,但是,这样的Poisson分布不能满足我们的要求,因此,我们引入一个先验,即:
不知道为什么要这样做,但是这时,问题可以描述为:
- 负二项分布
这时,可以证明X服从负二项分布(negative binomial distribution)。
这样的分布其实已经可以满足scHiC数据的离散特性和过度分散性。但是,实际的scHiC数据还具有稀疏性,因此在NB的基础上,进一步的发展出ZINB,即零膨胀负二项分布。
- 零膨胀负二项分布
定义一个示性函数$I_0(x)$:零膨胀负二项分布的概率密度函数为:
式中,$\theta$可以认为是真实的基因表达值被观测为0的概率。

