补充阅读一些关于ATAC的论文
snapATAC
1. introduction
The current analysis methods often require performing linear dimensionality reduction. In addition, the unsupervised identification of cell types or states in complex tissues using scATAC-seq dataset does not have the same degree of sensitivity as that from scRNA-seq.A sufficient number of single-cell profiles would be required to create robust aggregate signal for creating the peak reference.
To overcome these limitations, a software package, Single Nucleus Analysis Pipeline for ATAC-seq—SnapATAC is developed, that does not require population-level peak annotation prior to clustering.
2. results
input: binary cell-by-bin matrix with a resolution of 5kb
SnapATAC provides several useful features:
embedding
Nyström method:(1) it computes the low-dimension embedding for a subset of selected cells (also known as landmarks); (2) it projects the remaining cells to the embedding structure learned from the landmarks.- shortcoming:Nyström method is stochastic and could yield different clustering results in each sampling.
To overcome this limitation, a consensus approach is used that combines a mixture of low-dimensional manifolds learned from different sets of sampling.
- annotation to the clustering result
- indentification of the candidate regulatory elements
- link candidate regulatory elements to their putative target genes
- construct cellular trajectories from single-cell ATAC-seq
They compared the snapATAC methods with other published scATAC-seq analysis methods, which is quantified by both ARI and NMI. Also some experiments are performed to evaluate the robustness of snapATAC.
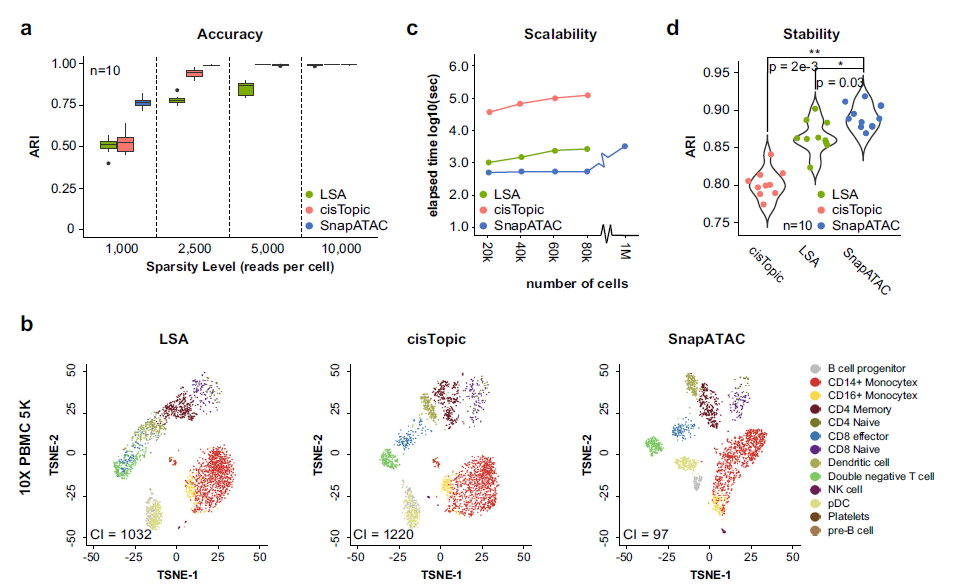
They thought that the improvment performence of snapATAAC comes from that it considers all reads from each cell.
3. discussion
- snapATAC is a useful tool for analyzing scATAC-seq data.
- snapATAC dosen’t require population-level peak annotation prior to clustering.
- snapATAC is applied to newly generated scATAC-seq datasets from mouse brain and human PBMCs and the results are consistent with the known cell types.
4. method
data processing:
The very origin input for snapATAC is the fastq files. The fastq files are processed by flloing steps: 1. add barcodes and demultiplexing, 2. reads alignment with bwa and sorted by reads name with samtools, 3. QC and reads filtering(don’t know anything about this part), 4. barcode filtering: filter cells by number of unique fragments and the fragments in promoter ratio, 5. bin filtering with blaklist from ENCODE, 6. binarization: convert the binary cell-by-bin matrix with a resolution of 5kb.
data file format: snap-file.
They carried experiments to different bin size matrix from 1kb to 10kb, and they choose 5kbas the default bin width for the analysis.
As for dimension reduction, they used the Nyström method to compute the low-dimension embedding for a subset of selected cells (also known as landmarks); it projects the remaining cells to the embedding structure learned from the landmarks. The number of landmarks is set to 1000 by default. The number of nearest neighbors is set to 15 by default(the proof process will be added in future).

